Μαρία Γιαβροπούλου1, Σοφία Βλάχου2 , Εύα Κασσή3
- Διευθύντρια ΕΣΥ
- Ακαδημαικός Υπότροφος, Ιατρική Σχολή, ΕΚΠΑ
- Αν.Καθηγήτρια, Ιατρική Σχολή, ΕΚΠΑ
Ενδοκρινολογική Μονάδα, Α’ Προπαιδευτική Παθολογική Κλινική, Γ.Ν.Α «ΛΑΙΚΟ», Κέντρο αναφοράς διαταραχών ασβεστίου-φωσφόρου
Οι πιο γνωστές και κοινές παθήσεις των οστών είναι η οστεoπενία-οστεοπόρωση, η οστεομαλακία από ανεπάρκεια της βιταμίνης D και ο πρωτοπαθής υπερπαραθυρεοειδισμός που οφείλεται σε αδένωμα των παραθυρεοειδών αδένων. H οστεοπενία – οστεοπόρωση εμφανίζονται κυρίως σε γυναίκες στην εμμηνόπαυση, η οστεομαλακία από ανεπάρκεια της βιταμίνης και στα 2 φύλα σε όλες τις ηλικίες όταν υπάρχει περιορισμένη έκθεση στον ήλιο (στα παιδιά χαρακτηρίζεται ως ραχίτιδα) και ο πρωτοπαθής υπερπαραθυρεοειδισμός μπορεί να είναι σποραδικός (αδένωμα) ή μέρος κληρονομικών συνδρόμων, κυρίως όταν εμφανίζεται πριν την ηλικία των 50 ετών.
Σπανιότερα και λιγότερο γνωστά νοσήματα των οστών και του μεταβολισμού ασβεστίου και φωσφόρου περιλαμβάνουν παθήσεις που χαρακτηρίζονται από:
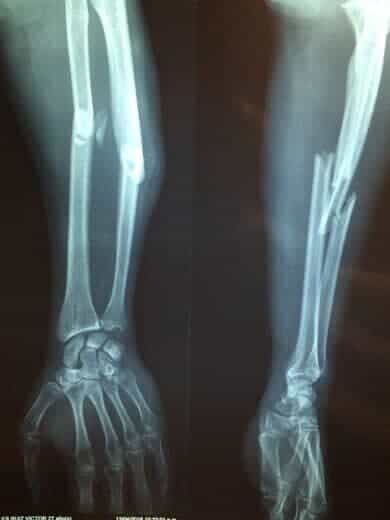
1) Aτελή ανάπτυξη και επιμετάλλωση των οστών οδηγώντας σε οστικές παραμορφώσεις και κατάγματα (πολλαπλά, ξεκινώντας από τη βρεφική ή την παιδική ηλικία, χαμηλής βίας δηλαδή χωρίς σοβαρό τραυματισμό, ανεξήγητα) (εικόνα 1).
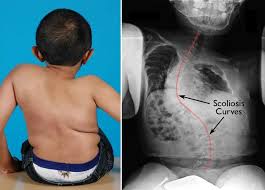
2) Ραχίτιδα (εικόνα 2) και σοβαρή σκολίωση.
3) Οστικά και διάχυτα μυϊκά άλγη
4) Διαταραχές οδοντοφυίας και ατελή οδοντογένεση ορατές ήδη από τη βρεφική ή παιδική ηλικία, πρόωρη απώλεια νεογιλών ή ενήλικων δοντιών, σοβαρή τερηδόνα.
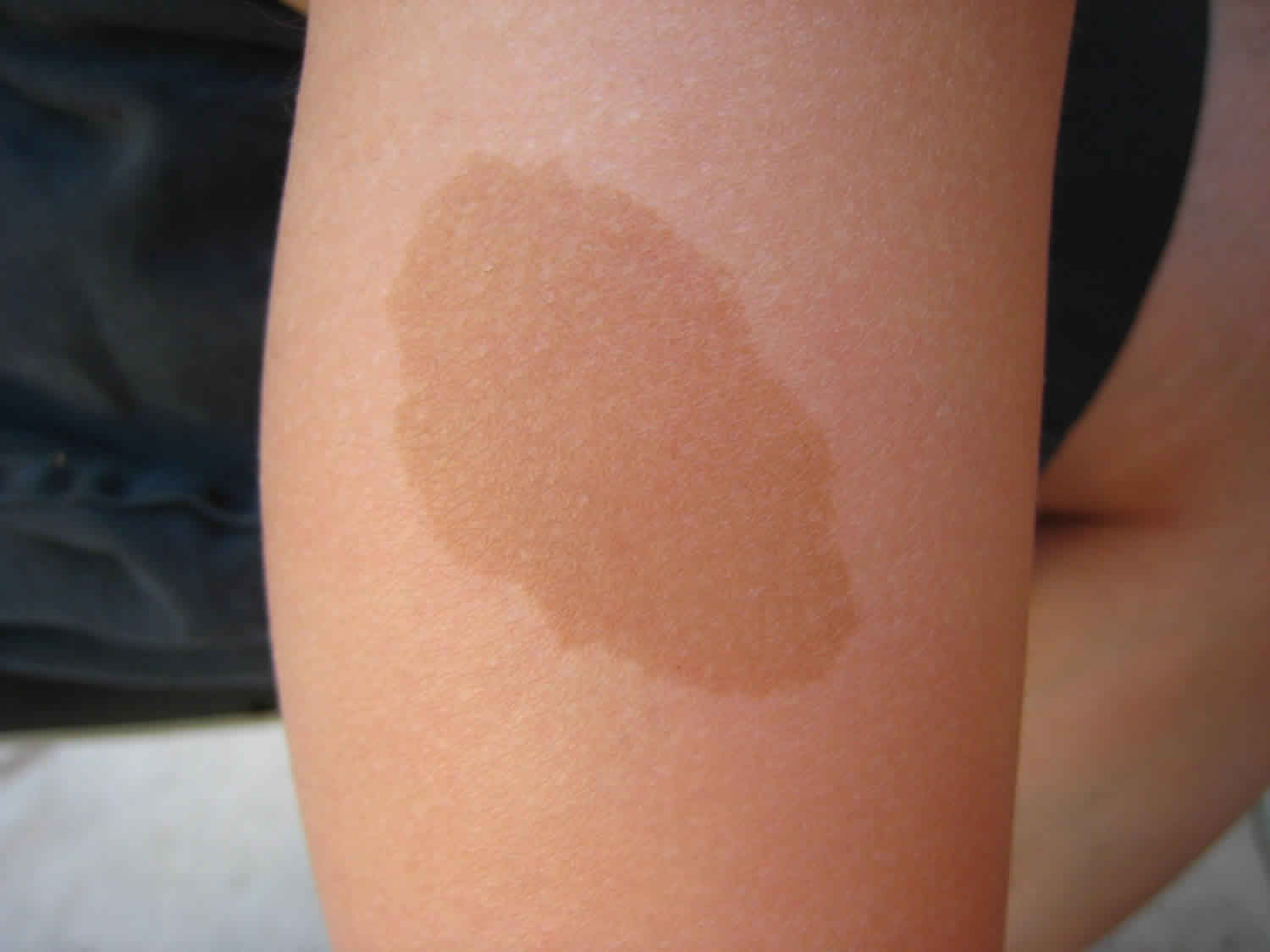
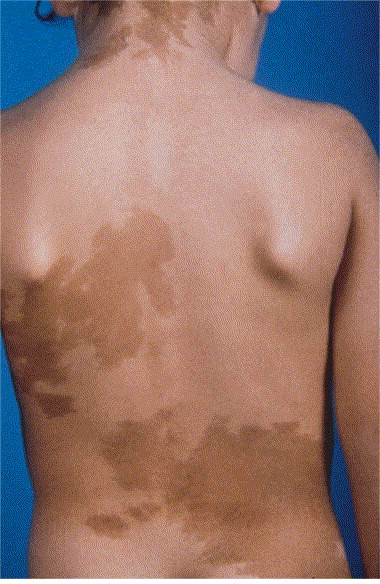
5) Δερματικές κηλίδες (περιοχές του δέρματος πιο σκούρου χρώματος από το υπόλοιπο δέρμα) ακανόνιστων ορίων (café-au-lait) (εικόνα 3).
6) Διαταραχές του χρώματος του σκληρού χιτώνα των ματιών (γκρι ή μπλε αντί για λευκό).
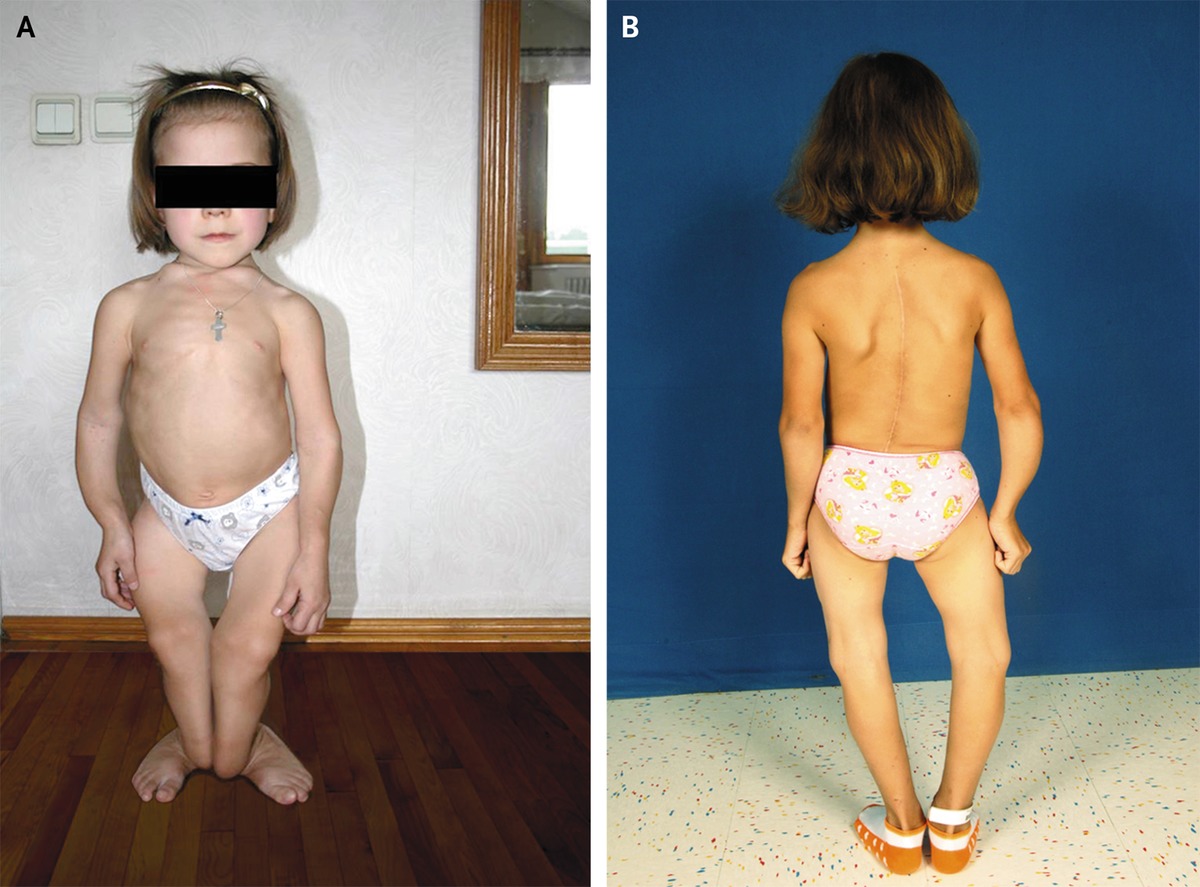
7) Διαταραχές σκελετικής ανάπτυξης όπως κοντό ανάστημα, βραχύς λαιμός, στρογγυλό πρόσωπο, κοντά δάχτυλα χεριών.
8) Κρανιοσυνοστέωση και απώλεια ακοής.
9) Διαταραχές του ασβεστίου με αυξημένα ή μειωμένα επίπεδα ασβεστίου στο αίμα οδηγώντας σε σχηματισμό λίθων ή διάχυτης ασβέστωσης στους νεφρούς και σε άλλα σημεία του σώματος, κωλικούς νεφρών, καρδιακές αρρυθμίες, νευροψυχιατρικές διαταραχές, επεισόδια τετανίας (μυϊκά τινάγματα και σπασμούς) και μυϊκή αδυναμία.
10) Διαταραχές του φωσφόρου με αυξημένα ή μειωμένα επίπεδα φωσφόρου στο αίμα με αποτέλεσμα το σχηματισμό συμπλόκων στο δέρμα και σε άλλους ιστούς, καρδιακές αρρυθμίες, μυική αδυναμία, ανεπάρκεια αναπνευστικών μυών και ραχίτιδα.
11) Εμφάνιση ενδοκρινοπαθειών όπως πρώϊμη εφηβεία, υπερλειτουργία του θυρεοειδούς αδένα, υπερέκκριση αυξητικής ορμόνης, προλακτίνης και κορτιζόλης, φαιοχρωμοκύττωμα.
12) Εμφάνιση άλλων όγκων όπως αδενώματα υπόφυσης και επινεφριδίων, νευροενδοκρινείς όγκοι γαστρεντερικού συστήματος, επασβεστώσεις και μυξώματα μαλακών μορίων.
Αξίζει να τονιστεί πως κάποιες από τις συγκεκριμένες παθήσεις είναι ήπιες και δεν χρειάζονται θεραπεία, άλλες όμως έχουν επιπλοκές, οξείες και χρόνιες και αν παραμείνουν αδιάγνωστες προκαλούν σημαντικά προβλήματα στην υγεία και την ποιότητα ζωής των ασθενών. Επιπλέον, η κληρονομική φύση των νοσημάτων αυτών στην πλειοψηφία τους μας οδηγεί στον γενετικό έλεγχο των συγγενών συμβάλλοντας με αυτόν τον τρόπο στην έγκαιρη διάγνωση και αντιμετώπιση της νόσου και την ανακούφιση των πασχόντων.
Τα σπανιότερα και λιγότερο γνωστά νοσήματα των οστών και του μεταβολισμού ασβεστίου και φωσφόρου είναι τα εξής:
Α] Σπάνιες διαταραχές του μεταβολισμού ασβεστίου και φωσφόρου
1.Οικογενής Καλοήθης Υπασβεστιουρική Υπερασβεστιαιμία
Η οικογενής καλοήθης υπασβεστιουρική υπερασβεστιαιμία είναι μια ασυμπτωματική γενετική διαταραχή του μεταβολισμού του ασβεστίου και χαρακτηρίζεται από μια ήπια υπερασβεστιαιμία με φυσιολογική ή μειωμένη νεφρική απέκκριση του ασβεστίου και ήπια αυξημένη ή φυσιολογική τιμή της παραθορμόνης. Οι ασθενείς είναι συνήθως ασυμπτωματικοί ενώ σπάνια μπορούν να εμφανισθούν συμπτώματα κόπωσης, αδυναμίας, υπερβολικής δίψας και απώλειας συγκέντρωσης. Η αντιμετώπιση της πάθησης αυτής συνίσταται σε μακροχρόνια παρακολούθηση και δεν χρήζει χειρουργικής παρέμβασης.
2. Πολλαπλή ενδοκρινική νεοπλασία τύπου 1 (ΜΕΝ1)
Το σύνδρομο πολλαπλής ενδοκρινικής νεοπλασίας τύπου 1 (ΜΕΝ1) χαρακτηρίζεται από την ανάπτυξη νευροενδοκρινών όγκων των παραθυρεοειδών αδένων, του παγκρέατος, και του προσθίου λοβού της υπόφυσης και λιγότερο συχνά του φλοιού των επινεφρίδιων και άλλων μη ενδοκρινικών όγκων. Σε ασθενείς με σύνδρομο ΜΕΝ1 οι όγκοι μπορούν να αναπτυχθούν σε οποιαδήποτε ηλικία και συνήθως το 95% των ασθενών εμφανίζουν κάποιον όγκο μέχρι την 5η δεκαετία της ζωής τους. Οι όγκοι των παραθυρεοειδών αδένων είναι συνήθεις και συχνά αποτελούν την πρώτη εκδήλωση του συνδρόμου.
Ο πρωτοπαθής υπερπαραθυρεοειδισμός οφείλεται στην υπερβολική έκκριση παραθορμόνης (PTH) από τους υπερπλαστικούς παραθυρεοειδείς αδένες. Οδηγεί σε αυξημένα επίπεδα ασβεστίου αίματος (υπερασβεστιαιμία) και αυξημένη αποβολή ασβεστίου από τα ούρα (υπερασβεστιουρία). Σπάνιες μορφές πρωτοπαθούς υπερπαραθυρεοειδισμού στα πλαίσια του συνδρόμου είναι αυτές που συναντάμε σε παιδιά και νεαρούς ενήλικες και οφείλονται σε κληρονομούμενες μεταλλάξεις γονιδίων. Τα συμπτώματα σχετίζονται με τα αυξημένα επίπεδα του ασβεστίου στο αίμα και τις επιπλοκές που σχετίζονται με το ουροποιητικό σύστημα (νεφροί, ουρητήρες) λόγω της ασβεστιουρίας καθώς και με συγκεκριμένα κλινικά χαρακτηριστικά ανάλογα με την γονιδιακή μετάλλαξη (φαινότυπος γενετικού συνδρόμου).
3. Πολλαπλή ενδοκρινική νεοπλασία τύπου 2 (ΜΕΝ2)
Το σύνδρομο πολλαπλής ενδοκρινικής νεοπλασίας τύπου 2 (Multiple EndocrineNeoplasia type2, ΜΕΝ2) χαρακτηρίζεται από την εμφάνιση μυελοειδούς καρκίνου του θυρεοειδούς αδένα, φαιοχρωμοκυττώματος και στον έναν τύπο του συνδρόμου από την εμφάνιση πρωτοπαθούς υπερπαραθυρεοειδισμού. Τα κλινικά χαρακτηριστικά του ΜΕΝ 2 σχετίζονται από τον υπότυπο (2α ή 2β ).
4. Νεοπλάσματα Παραθυρεοειδών Αδένων
Ο καρκίνος των παραθυρεοειδών αδένων, συνήθως εμφανίζεται με σοβαρή υπερασβεστιαιμία, ανορεξία, πολυουρία, κόπωση, αφυδάτωση, οστικά άλγη, υποπεριοστική οστική απορρόφηση, παθολογικά κατάγματα και επιπλοκές στο ουροποιητικό σύστημα λόγω της υπερασβεστιουρίας.
5. Υποπαραθυρεοειδισμός
Ο υποπαραθυρεοειδισμος οφείλεται στη μειωμένη παραγωγή/ ή έκκριση της παραθορμόνης από τους παραθυρεοειδείς αδένες. Χαρακτηρίζεται από χαμηλά επίπεδα ασβεστίου, υψηλή τιμή φωσφόρου και πολύ χαμηλή τιμή παραθορμόνης. Τα συμπτώματα οφείλονται κυρίως στα χαμηλά επίπεδα ασβεστίου και μπορεί να περιλαμβάνουν παραισθησίες, μουδιάσματα (κυρίως γύρω από το στόμα και στα δάχτυλα των άνω άκρων), επιληπτικές κρίσεις και κλινικά σημεία τετανίας (μυϊκά τινάγματα και σπασμοί κάτω και άνω άκρων). Σε σοβαρές περιπτώσεις μπορεί να εμφανιστούν καρδιακές επιπλοκές. Οι μακροχρόνιες επιπλοκές της νόσου είναι αρκετά συχνές και περιλαμβάνουν έκτοπη επασβέστωση των ιστών, νεφρασβέστωση, γνωσιακές και νευροψυχιατρικές διαταραχές. Στα παιδιά ο υποπαραθυρεοειδισμός οφείλεται κυρίως σε ανωμαλίες στη διάπλαση των παραθυρεοειδών αδένων ενώ σε μεγαλύτερες ηλικίες μπορεί να οφείλεται σε αυτοάνοση καταστροφή των παραθυρεοειδών αδένων, παρενέργεια φαρμακευτικής αγωγής ή χειρουργείου στην περιοχή του τραχήλου (πχ. Θυρεοειδεκτομή).
6. Σύνδρομα Ψευδοϋποπαραθυρεοειδισμού
Ο όρος ψευδοϋποπαραθυρεοειδισμός περιγράφει μια ομάδα νοσημάτων με εκδηλώσεις παρόμοιες με αυτές του υποπαραθυρεοειδισμού (εκδηλώνεται με υπασβεστιαιμία και υπερφωσφοραιμία) ο οποίος όμως οφείλεται σε αντίσταση των ιστών–στόχων στην δράση της παραθορμόνης. Οι ασθενείς με ψευδοϋποπαραθυρεοειδισμό μπορεί να χαρακτηρίζονται επιπλέον και από διάφορα άλλα κλινικά χαρακτηριστικά όπως στρογγυλό προσωπείο, παχυσαρκία, βραχυσωμία, βραχυδακτυλία, έκτοπη επασβέστωση των ιστών που συνολικά περικλείονται στον όρο κληρονομική οστεοδυστροφία Albright.
Η ακριβής επίπτωση του ψευδοϋποπαραθυρεοειδισμού είναι άγνωστη. Ο υποκείμενος παθοφυσιολογικός μηχανισμός της νόσου σχετίζεται με γενετικές ή επιγενετικές αλλαγές στα γονίδια που εμπλέκονται στο ενδοκυττάριο σηματοδοτικό μονοπάτι της παραθορμόνης.
Η διάγνωση βασίζεται στην παρουσία των κλινικών και των εργαστηριακών ευρημάτων που είναι χαρακτηριστικά για την νόσο.
Γενετική συμβουλευτική: Ο ψευδοϋποπαραθυρεοειδισμός μπορεί να είναι σποραδικός ή να κληρονομείται με τον αυτοσωματικό επικρατούντα χαρακτήρα. Στις κληρονομικές περιπτώσεις συνιστάται γενετική συμβουλευτική.
Η αποτελεσματική αντιμετώπιση των ασθενών με ψευδοϋποπαραθυρεοειδισμό χρήζει πολυεπιστημονικής προσέγγισης με ιατρούς διαφόρων ειδικοτήτων καθώς και συγκεκριμένες παρεμβάσεις. Η αντιμετώπιση συνίσταται σε γενικά μέτρα (συμβουλές για διατροφή και δίαιτα προς αποφυγή της παχυσαρκίας, και φυσιοθεραπεία) και ειδικά μέτρα που στοχεύουν στην διόρθωση της υπασβεστιαιμίας, και των άλλων ενδοκρινολογικών διαταραχών καθώς και σε εστιασμένες θεραπείες όπως ορθοπεδική αποκατάσταση της σπονδυλικής στήλης και οδοντιατρική φροντίδα.
Η πρόγνωση διαφέρει κατά περίπτωση ανάλογα με την ηλικία εμφάνισης και τη σοβαρότητα των συμπτωμάτων της νόσου.
7. Κληρονομικές μορφές Υποφωσφοραιμικής Ραχίτιδας
Η ανεπάρκεια φωσφόρου οδηγεί σε υποφωσφοραιμία και ως εκ τούτου σε οστεομαλακία, ραχίτιδα (στα παιδιά), διαταραχές επιμετάλλωσης των οστών, οστικές δυσμορφίες, οστικά άλγη, καθυστερημένη σκελετική ανάπτυξη, ανωμαλίες οδόντων και μυών. Επιπλέον χαρακτηριστικά που μπορούν να εμφανιστούν είναι η νεφρασβέστωση, η κρανιοσυνοστέωση, η απώλεια ακοής, οι ενθεσοπάθειες και ο δευτεροπαθής υπερπαραθυρεοειδισμός. Υποφωσφοραιμία η οποία οφείλεται σε νεφρική απώλεια του φωσφόρου είναι αποτέλεσμα γενετικών διαταραχών που οδηγούν σε αυξημένη παραγωγή ή δράση του φωσφατουρικού ινοβλαστικού αυξητικού παράγοντα FGF23. Σε σπάνιες περιπτώσεις η διαταραχή των επίπεδων του φωσφόρου οφείλεται σε ανωμαλίες κατά την νεφρική διαχείρισή του (γενετικές ή επίκτητες).
8. Επίκτητη ογκογενής υποφωσφοραιμία – Σύνδρομα Ογκογενούς Οστεομαλακίας
Η ογκογενής υποφωσφοραιμία ή ογκογενής οστεομαλακία οδηγεί σε σοβαρή υποφωσφοραιμία και νεφρική απώλεια φωσφόρου εξαιτίας υπερέκκρισης του φωσφατουρικού ινοβλαστικού αυξητικού παράγοντα FGF23 από μεσεγχυματικής προέλευσης όγκους. Τα επίπεδα ασβεστίου είναι συνήθως χαμηλά. Οι ασθενείς (συνήθως ενήλικες) παρουσιάζονται με σοβαρά μυϊκά προβλήματα, κατάγματα ευθραυστότητας, ψευδοκατάγματα, οστικά και διάχυτα μυϊκά άλγη. Για την σωστή αντιμετώπιση της νόσου είναι κριτικής σημασίας η εντόπιση του όγκου που υπερεκκρίνει FGF23 με την χρήση των καλυτέρων διαθέσιμων απεικονιστικών τεχνικών.
Τα τελευταία χρόνια είναι διαθέσιμη ειδική θεραπεία για τα υποφωσφοραιμικά σύνδρομα που στοχεύει ακριβώς στον αιτιολογικό παράγοντα, δηλαδή την αυξημένη έκκριση του ινοβλαστικού αυξητικού παράγοντα FGF-23. Η μπουροσουμάμπη (burosumab) είναι ένα ανασυνδυασμένο ανθρώπινο μονοκλωνικό αντίσωμα (IgG1), το οποίο δεσμεύεται στον αυξητικό παράγοντα FGF23 και αναστέλλει τη δράση του. Αναστέλλοντας τον FGF23, η μπουροσουμάμπη αυξάνει τη σωληναριακή επαναρρόφηση φωσφόρου από τον νεφρό και αυξάνει τη συγκέντρωση της 1, 25 διϋδροξυ-βιταμίνης D στον ορό.
9. Υπερφωσφοραιμία και ογκογενές σύνδρομο έκτοπων επασβεστώσεων
Η υπερφωσφοραιμία μπορεί να οφείλεται σε διαταραχές της νεφρικής απέκκρισης φωσφόρου.Οι περισσότερες περιπτώσεις έχουν γενετικό υπόβαθρο και χαρακτηρίζονται από έλλειψη του παράγοντα FGF23. Οι ασθενείς μπορούν να παρουσιάσουν πόνο στα οστά και τις αρθρώσεις,και συχνά σοβαρές έκτοπες ασβεστώσεις ιστών και μαλακών μορίων.
Β] Σπάνια νοσήματα διαταραχών του οστικού μεταβολισμού
1. Ινώδης δυσπλασία – Σύνδρομο Mc Cune – Albright
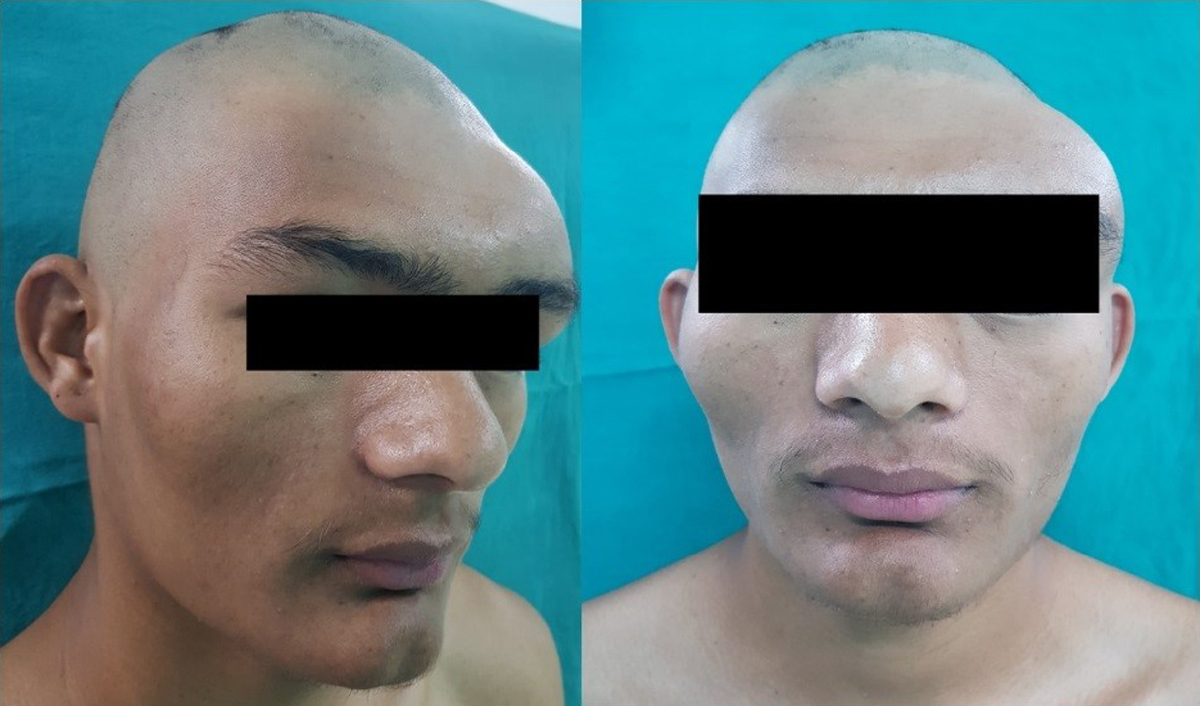
Ο ακριβής επιπολασμός της ινώδους δυσπλασίας των οστών είναι άγνωστος, αλλά είναι μικρότερος από 1: 2000. Το σύνδρομο MacCune Albright είναι μια σπάνια διαταραχή που χαρακτηρίζεται από σκελετικές αλλοιώσεις, μελάγχρωση/υπερχρωματισμό του δέρματος και υπερ-λειτουργικές ενδοκρινοπάθειες. Η συγκεκριμένη νόσος αποτελεί ένα μωσαϊκό κλινικών εκδηλώσεων με ευρύ φάσμα, που κυμαίνεται από ένα τυχαίο ακτινολογικό εύρημα έως σοβαρή νόσο που προκαλεί αναπηρία. Η διάγνωση των διαφορετικών μορφών της ινώδους δυσπλασίας/σ.MacCune Albright μπορεί να γίνει μόνο μετά από ενδελεχή κλινική αξιολόγηση για α) την έκταση της σκελετικής νόσου: μονοοστική / πολυοστική και β) την παρουσία εξωσκελετικών εκδηλώσεων. Η μονο-οστική ινώδης δυσπλασία η βλάβη περιορίζεται σε ένα οστό, ενώ στην πολυοστική μορφή της νόσου η βλάβη εντοπίζεται σε περισσότερες από μία σκελετικές θέσεις (εικόνα 4).
Το σύνδρομο Mc Cune – Albright ορίζεται ως ο συνδυασμός της ινώδους δυσπλασίας και μίας ή περισσότερων εξωσκελετικών εκδηλώσεων ή παρουσία δύο ή περισσότερων εξωσκελετικών ευρημάτων
Το σύνδρομο Mazabraud είναι ο συνδυασμός ινώδους δυσπλασίας των οστών με μυξώματα μαλακών μορίων. Το μυξώμα ορίζεται ως έξω-σκελετική εκδήλωση της ινώδους δυσπλασίας και μπορεί να συμβεί σε συνδυασμό με οποιονδήποτε τύπο της νόσου (μονοοστικό, πολυοστικό ή σύνδρομο MacCune Albright). Άλλα εξωσκελετικά χαρακτηριστικά περιλαμβάνουν: α) τις δερματικές κηλίδες café-au-lait με χαρακτηριστική μορφολογία ακανόνιστων ορίων (Coast of Maine) με κατανομή που περιορίζεται στη μία πλευρά του σώματος (εικόνα 5) και β) διαταραχές πολλών ενδοκρινών αδένων. Η ανεξάρτητη από τις γοναδοτροπίνες αυξημένη παραγωγή των ορμονών του φύλου οδηγεί σε πρώιμη εφηβεία, υποτροπιάζουσες κύστεις των ωοθηκών σε κορίτσια και γυναίκες ή αυτόνομη παραγωγή τεστοστερόνης στα αγόρια και στους άντρες. Παρατηρούνται επίσης βλάβες των όρχεων, υπερλειτουργία του θυρεοειδούς αδένα, υπερέκκριση αυξητικής ορμόνης και υπερκορτιζολαιμία-σύνδρομο Cushing. Αξίζει να σημειωθεί ότι η υποφωσφοραιμία που σχετίζεται με τον παράγοντα FGF-23 δεν θεωρείται χαρακτηριστικό του συνδρόμου McCune-Albright αλλά αποτελεί μάλλον δείκτη της σοβαρότητας της σκελετικής νόσου (ινώδης δυσπλασία).
Ενώ πρόκειται για μια γενετική νόσο, η μετάλλαξη εμφανίζεται μετά τη γονιμοποίηση και έτσι δεν κληρονομείται. Προγεννητική διάγνωση δεν εφαρμόζεται. Η θεραπεία περιλαμβάνει γενικά μέτρα (παροχή πληροφοριών σχετικά με την ασθένεια, συμβουλές για τον τρόπο ζωής, άσκηση και φυσιοθεραπεία σε περίπτωση καταγμάτων) και συγκεκριμένα μέτρα, όπως η αντιμετώπιση της επαγόμενης από τον FGF-23 νεφρικής απώλειας φωσφόρου, της σκολίωσης, του οστικού πόνου, του συνδρόμου Mazabraud, των συνοδών ενδοκρινοπαθειών, των αιματολογικών εκδηλώσεων, των γαστρεντερικών εκδηλώσεων και του αυξημένου κινδύνου κακοήθειας. Η χειρουργική αντιμετώπιση περιλαμβάνει ορθοπεδική, νευροχειρουργική, κρανιοπροσωπική, γναθοχειρουργική και οδοντιατρική παρέμβαση. Η πρόγνωση εξαρτάται από τη σοβαρότητα της νόσου και την ανταπόκριση στη φαρμακευτική και τη χειρουργική θεραπεία.
2. Ατελής Οστεογένεση
Η ατελής οστεογένεση (ΑΟ) (osteogenesis imperfecta,) περιλαμβάνει μια ετερογενή ομάδα γενετικών διαταραχών που χαρακτηρίζονται από αυξημένη ευθραυστότητα των οστών, χαμηλή οστική μάζα και αυξημένο κίνδυνο οστικών καταγμάτων χαμηλής βίας.
Ο επιπολασμός της νόσου εκτιμάται μεταξύ 1 / 10.000 και 1 / 20.000.
Η ηλικία κατά τη διάγνωση εξαρτάται από τη σοβαρότητα της νόσου. Πέντε κλινικά διακριτές μορφές της νόσου έχουν αναγνωριστεί. Η πιο κοινή και χαρακτηριστική κλινική εκδήλωση όλων των μορφών ΑΟ είναι η ευθραυστότητα των οστών, η οποία εκδηλώνεται ως πολλαπλά αυτόματα κατάγματα.
Η διάγνωση βασίζεται στα ειδικά σκελετικά και εξωσκελετικά κλινικά ευρήματα. Χαρακτηριστικό κλινικό σημείο αποτελεί το σκούρο χρώμα (μπλε, γκρι) του φυσιολογικά λευκού σκληρού χιτώνα των οφθαλμών (εικόνα 6).

Η υποψία της νόσου μπορεί να τεθεί προγεννητικά από το υπερηχογράφημα και / ή να επιβεβαιωθεί μέσω της μοριακής ανάλυσης σε αμνιοκύτταρα ή κύτταρα χοριακών λαχνών αν έχει εντοπιστεί η υπεύθυνη μετάλλαξη στην οικογένεια.
Η αντιμετώπιση είναι πολύπλευρη με τη συμμετοχή έμπειρων ιατρών διαφόρων ειδικοτήτων (ενδοκρινολόγων, ορθοπεδικών, φυσιάτρων, φυσικοθεραπευτών και φυσιάτρων ειδικών αποκατάστασης). Τα διφωσφονικά, λόγω της ισχυρής αντι-οστεοκλαστικής τους ιδιότητας, είναι η αγωγή πρώτης επιλογής αλλά δεν συνιστούν ειδική θεραπεία. Η πρόληψη της ανεπάρκειας βιταμίνης D και ασβεστίου στους ασθενείς αυτούς είναι απαραίτητη καθ ‘όλη τη διάρκεια της ζωής.
Η χειρουργική παρέμβαση είναι απαραίτητη για τη διόρθωση των παραμορφώσεων των οστών και της σπονδυλικής στήλης και την πρόληψη των καταγμάτων των μακρών οστών (κεντρο-μυελική οστεοσύνθεση). Η έγκαιρη έναρξη φυσικοθεραπείας βελτιώνει την αυτονομία των ασθενών βοηθώντας στην αξιολόγηση τυχόν κινητικών ελλειμμάτων, μειώνοντας τον κίνδυνο πτώσεων και ενθαρρύνοντας τους ασθενείς για αθλητική δραστηριότητα.
Η πρόγνωση της λειτουργικότητας εξαρτάται από τη σοβαρότητα της νόσου και από την ποιότητα της αντιμετώπισης. Το προσδόκιμο επιβίωσης εξαρτάται από τη σοβαρότητα των παραμορφώσεων της σπονδυλικής στήλης (εικόνα 7) λόγω των αναπνευστικών επιπλοκών που προκύπτουν από αυτές.
3. Υποφωσφατασία
Η υποφωσφατασία είναι μια σπάνια κληρονομική μεταβολική διαταραχή που χαρακτηρίζεται από ελαττωματική επιμετάλλωση οστών και / ή δοντιών παρουσία μειωμένης δραστηριότητας του ενζύμου αλκαλικής φωσφατάση.
Το κλινικό φάσμα είναι εξαιρετικά ευρύ, από ενδομήτριο θάνατο έως κατάγματα των κάτω άκρων στην ενήλικο ζωή, ή ακόμη και απουσία οστικής εκδήλωσης (οδοντοϋποφωσφατασία).
Δεν υπάρχουν διαθέσιμα ακριβή δεδομένα για τον επιπολασμό και τη συχνότητα εμφάνισης της υποφωσφατασίας. Ο επιπολασμός των σοβαρών μορφών της νόσου εκτιμάται σε 1 /300.000 γεννήσεις στην Ευρώπη.
Η κλινική εικόνα της μορφής της νόσου με έναρξη στην παιδική ηλικία κυμαίνεται από χαμηλή οστική πυκνότητα με ανεξήγητα κατάγματα έως την εμφάνιση ραχίτιδας. Η ενήλικη μορφή της νόσου συνεπάγεται πρόωρη απώλεια τελικών οδόντων και κατάγματα χαμηλής βίας των κάτω άκρων κατά τη διάρκεια της μέσης ηλικίας.
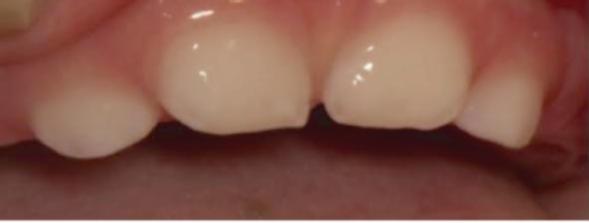
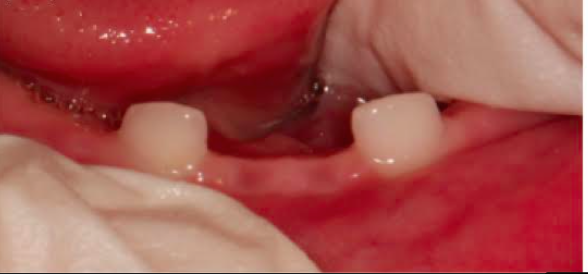
Τέλος, η οδοντοϋποφωσφατασία περιλαμβάνει πρόωρη απώλεια νεογιλών δοντιών και / ή σοβαρή τερηδόνα (εικόνες 8, 9). Σπάνιες περιπτώσεις βρεφών με υποφωσφατασία που έχουν φυσιολογική δράση αλκαλικής φωσφατάσης στον ορό θεωρούνται ότι πάσχουν από «ψευδοϋποφωσφατασία».
Η διάγνωση βασίζεται σε εργαστηριακές εξετάσεις και μοριακούς γενετικούς ελέγχους του γονιδίου ALP για την ανίχνευση αιτιολογικών γενετικών μεταλλάξεων. Η δραστηριότητα της αλκαλικής φωσφατάσης στον ορό είναι σημαντικά μειωμένη ενώ η φωσφοαιθανολαμίνη των ούρων (PEA) είναι αυξημένη αλλά οι διαταραχές αυτές δεν είναι παθογνωμονικές.
Το υπερηχογράφημα είναι χρήσιμο για τη διάγνωση της προγεννητικής και περινεογνικής μορφής της νόσου. Η κλινική εξέταση και ο ακτινολογικός έλεγχος συμβάλλουν στη διάγνωση της βρεφικής, παιδικής και ενήλικης μορφής της νόσου.
Η προγεννητική διάγνωση γίνεται με γενετική ανάλυση σε κύτταρα χοριακών λαχνών για την αναζήτηση της μετάλλαξης.
Γενετική συμβουλευτική: Η περινεογνική και σοβαρή βρεφική μορφή HPP κληρονομούνται σύμφωνα με τον αυτοσωματικό υπολειπόμενο πρότυπο κληρονομικότητας. Ο προγεννητικός καλοήθης τύπος της νόσου, η μέτριας βαρύτητας βρεφική μορφή της νόσου, η ενήλικη μορφή της νόσου και η οδοντοϋποφωσφατασία κληρονομούνται με τον αυτοσωματικό υπολειπόμενο ή κυρίαρχο τύπο, ανάλογα με την επίδραση της μετάλλαξης στην δραστικότητα του ενζύμου της αλκαλικής φωσφατάσης. Οι λιγότερο σοβαρές μορφές της νόσου είναι πιθανότερο να κληρονομούνται με τον επικρατούντα χαρακτήρα. Η ποικιλία στον τρόπο κληρονομικότητας εξηγεί εν μέρει την κλινική ετερογένεια.
Η υποστηρικτική συμπτωματική θεραπεία σε παιδικές και ενήλικες μορφές περιλαμβάνει μη στεροειδή αντιφλεγμονώδη φάρμακα (παιδιά), οστεοαναβολική αγωγή (τεριπαρατίδη) (ενήλικες) και ορθοπεδική παρέμβαση. Τα τελευταία χρόνια είναι διαθέσιμη και ειδική θεραπεία με υποκατάσταση του ενζύμου (αλκαλική φωσφατάση) που λείπει από του ασθενείς (ασφωτάση άλφα) με πολύ ικανοποιητικά αποτελέσματα κυρίως στα παιδιά αλλά και στους ενήλικες που εμφανίζουν σοβαρή συμπτωματολογία.
Η τακτική παρακολούθηση και η κατάλληλη υγιεινή και φροντίδα των δοντιών είναι απαραίτητες.
Η περινεογνική μορφή είναι σχεδόν πάντα θανατηφόρα μέσα σε ημέρες ή εβδομάδες. Αναπνευστικές επιπλοκές οδηγούν σε υψηλά ποσοστά θνησιμότητας στη βρεφική μορφή. Το προσδόκιμο ζωής δεν θεωρείται ότι επηρεάζεται στην ενήλικη μορφή της νόσου ή στην οδοντοϋποφωσφατασία.
Από τη διάγνωση στην εξειδικευμένη αντιμετώπιση: Λίγα λόγια για το Εθνικό Κέντρο Εμπειρογνωμοσύνης σπανίων νοσημάτων διαταραχών ασβεστίου και φωσφόρου
Στην προσπάθεια να υπάρχει μία συντονισμένη παρακολούθηση και ανταπόκριση στις ανάγκες των ασθενών με τα σπάνια αυτά νοσήματα η Ενδοκρινολογική μονάδα της Α΄ Προπαιδευτικής Παθολογικής Κλινικής του Πανεπιστημίου Αθηνών οργάνωσε ένα εξειδικευμένο κέντρο αναφοράς με πολυεπιστημονική εκπροσώπηση και παροχή εξειδικευμένης φροντίδας στου ασθενείς αυτούς.
Το Κέντρο Εμπειρογνωμοσύνης Σπανίων Ενδοκρινολογικών Νοσημάτων με εστίαση στις Διαταραχές Μεταβολισμού Ασβεστίου και Φωσφόρου έχει ως αντικείμενο ακριβώς τα σπάνια αυτά νοσήματα και την αντιμετώπισή τους. Στο πλαίσιο παροχής εξειδικευμένων υπηρεσιών από πρωτοπόρους στο γνωστικό τους αντικείμενο συμμετέχουν ιατροί και βιολόγοι και από άλλα Νοσοκομεία Πανεπιστημιακά ή μη δημιουργώντας μία πολυεπιστημονική ομάδα με ευρεία εκπροσώπηση όλων των εμπλεκόμενων με τα νοσήματα αυτά ειδικοτήτων.
Οι υπηρεσίες περιλαμβάνουν την έγκαιρη και ακριβή διάγνωση (συμπεριλαμβανομένων των γενετικών και μοριακών αναλύσεων), την παροχή εξειδικευμένης φροντίδας, , την επιλογή της κατάλληλης θεραπείας και τη συμβουλευτική προς τους ασθενείς και τους συγγενείς τους.
Συνολικά παρακολουθούνται στο Κέντρο περισσότεροι από 200 ασθενείς με σπάνιες διαταραχές μεταβολισμού ασβεστίου και φωσφόρου καθώς και σπάνια οστικά μεταβολικά νοσήματα.
Το Κέντρο είναι οργανωμένο έτσι ώστε να περιλαμβάνει όλες τις εμπλεκόμενες ειδικότητες σύμφωνα με τα προαπαιτούμενα του European Reference Network for Rare Endocrine Diseases of Calcium and Phosphate Metabolism (Ενδοκρινολογία, Παιδοενδοκρινολογία, Ορθοπαιδική, Ογκολογία, Χειρουργική, Πυρηνική Ιατρική, Ακτινοδιαγνωστική, Παθολογική Ανατομική και Γενετική). Επιπλέον, το Kέντρο περιλαμβάνει στην υποδομή του δευτερογενείς ειδικότητες στις οποίες ανήκουν η Καρδιολογία, Καρδιοχειρουργική, η Ψυχιατρική και η Διαιτολογία.
Όλα τα τμήματα που συμμετέχουν στο Κέντρο ακολουθούν τυποποιημένες διαδικασίες λειτουργίας (Standard Operating Procedures-SOPs).
Το Κέντρο συμμετέχει επίσης στη διεξαγωγή εθνικών μελετών σχετικών με τα σπάνια νοσήματα διαταραχών Ασβεστίου και Φωσφόρου και έχει δημοσιεύσει πλήθος πρωτότυπων μελετών και ανασκοπήσεων σε διεθνή κι ελληνικά επιστημονικά περιοδικά.
Tα Εξωτερικά Ιατρεία του Κέντρου Εμπειρογνωμοσύνης στεγάζονται σε έναν άρτια εξοπλισμένο, ενιαίο, και πρόσφατα ανακαινισμένο χώρο στο παράρτημα του Γ.Ν.Α«ΛΑΙΚΟ» στη Σεβαστουπόλεως 16 στο Γουδί, στον 5 όροφο, έτσι ώστε να εξασφαλίζεται η άμεση συνεργασία διαφόρων ιατρικών ειδικοτήτων όταν χρειάζεται, χωρίς περιττές μετακινήσεις και άσκοπες καθυστερήσεις.
Υποστηρίζονται από τα πλήρως εξοπλισμένα Εργαστήρια του Γ.Ν.Α «ΛΑΙΚΟ»: Κεντρικά Βιοχημικά Εργαστήρια, Παθολογοανατομικό Εργαστήριο, Ακτινολογικό Εργαστήριο, Τμήμα Πυρηνικής Ιατρικής.
Οι διαγνωστικές εξετάσεις των ασθενών περιλαμβάνουν κατά βάση αιματολογικό και βιοχημικό έλεγχο αίματος και ούρων, απεικονιστικές εξετάσεις (απλές ακτινογραφίες, αξονική-μαγνητική τομογραφία, σπινθηρογράφημα οστών ή σπινθηρογράφημα με sestamibi) και πιθανόν επεμβατικές εξετάσεις (βιοψία οστού). Περαιτέρω εξετάσεις που μπορεί να ζητηθούν αποτελούν ανασκόπηση ιστοπαθολογικών δειγμάτων, διενέργεια ειδικών σπινθηρογραφημάτων όπως 68Ga-PET/CT και όταν απαιτείται και 18FDG-PET/CT και μοριακός (γενετικός) έλεγχος.
Καθώς η εξειδικευμένη γνώση αναπτύσσεται συνεχώς με ταχείς ρυθμούς η κατάλληλη και εμπεριστατωμένη αντιμετώπιση σπανίων νοσημάτων αποτελεί μία άμεση πρόκληση για ολόκληρη την ιατρική επιστημονική κοινότητα. Η δημιουργία εξειδικευμένων Εθνικών Κέντρων Αναφοράς που συμμετέχουν στα αντίστοιχα Ευρωπαϊκά Δίκτυα Αναφοράς απαντάει ακριβώς σε αυτήν την πρόκληση για καλύτερη και πληρέστερη ιατρική κάλυψη των ασθενών αυτών.
Στο πλαίσιο αυτό, υπάρχει πλέον η δυνατότητα απευθείας επικοινωνίας των ασθενών αλλά και των θεράποντων ιατρών τους με το Κέντρου Εμπειρογνωμοσύνης του ΓΝΑ «ΛΑΙΚΟ» για οποιαδήποτε υποστήριξη που αφορά στη διάγνωση και σωστή αντιμετώπιση του νοσήματος.
Επικοινωνία
Γραμματεία
Διεύθυνση:Mιχαλακοπούλου 176, 11527, Γουδί, Αθήνα, Τηλ.Επικοινωνίας: 2107462217, E-mail:[email protected]
Γενικό Νοσοκομείο Αθηνών ΛΑΙΚΟ
Διεύθυνση:Αγίου Θωμά 17 11527, Γουδί, Αθήνα. Τηλ.Ραντεβού: 1535
Εξωτερικά Ιατρεία Κέντρου Εμπειρογνωμοσύνης (Κέντρο αναφοράς διαταραχών ασβεστίου-φωσφόρου: Σεβαστουπόλεως 16, 11527, Γουδί, Αθήνα, 5ος όροφος, Γραφείο Νο.5 (Δευτέρα – Τετάρτη, Υπεύθυνες : κα. Ε. Κασσή, κα Μ.Γιαβροπούλου)
Βιβλιογραφία
- Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, Ninth Edition.
- Taxonomy of rare genetic metabolic bone diseases. Masi L, Agnusdei D, Bilezikian J, Chappard D, Chapurlat R, Cianferotti L, Devolgelaer JP, El Maghraoui A, Ferrari S, Javaid MK, Kaufman JM, Liberman UA, Lyritis G, Miller P, Napoli N, Roldan E, Papapoulos S, Watts NB, Brandi ML. Osteoporos Int. 2015;26(10):2529-58.
- Rare bone diseases and their dental, oral and craniofacial manifestations. Foster BL, Ramnitz MS, Gafni RI, Burke AB, Boyce AM, Lee JS, Wright JT, Akintoye SO, Somerman MJ, Collins MT. J Dent Res. 2014 Jul;93(7 Suppl):7S-19S.
- Profile of asfotase alfa in the treatment of hypophosphatasia: design, development, and place in therapy. Foster B, Bowden S. Drug Des Devel Ther 2018;12:3147-3161.
- Pharmacodynamics of asfotase alfa in adults with pediatric-onset hypophosphatasia. Seefried L, Kishnani P, Moseley S , Denker A, Watsky E, Whyte M, Dahir K. Bone. 2020 Sep 26;115664.
- Burosumab in X-linked hypophosphatemia: a profile of its use in the USA. Lyseng-Williamson K. Drugs Ther Perspect. 2018; 34(11): 497–506
- Clinical Evidence for the Benefits of Burosumab Therapy for X-Linked Hypophosphatemia (XLH) and Other Conditions in Adults and Children. Schindeler A, Biggin A, Munns C. Front Endocrinol (Lausanne). 2020 May 28;11:338.